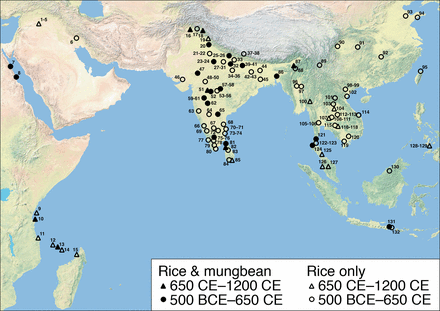
Figure S3 from the paper cited below.
Ancient evidence of Asian crops in Madagascar provide another piece of evidence for discerning the details of this long distance mass migration of Austronesian people to Africa.
Significance
The prehistoric settlement of Madagascar by people from distant Southeast Asia has long captured both scholarly and public imagination, but on the ground evidence for this colonization has eluded archaeologists for decades. Our study provides the first, to our knowledge, archaeological evidence for an early Southeast Asian presence in Madagascar and reveals that this settlement extended to the Comoros. Our findings point to a complex Malagasy settlement history and open new research avenues for linguists, geneticists, and archaeologists to further study the timing and process of this population movement. They also provide insight into early processes of Indian Ocean biological exchange and in particular, Madagascar’s floral introductions, which account for one-tenth of its current vascular plant species diversity.
Abstract
The Austronesian settlement of the remote island of Madagascar remains one of the great puzzles of Indo-Pacific prehistory. Although linguistic, ethnographic, and genetic evidence points clearly to a colonization of Madagascar by Austronesian language-speaking people from Island Southeast Asia, decades of archaeological research have failed to locate evidence for a Southeast Asian signature in the island’s early material record. Here, we present new archaeobotanical data that show that Southeast Asian settlers brought Asian crops with them when they settled in Africa. These crops provide the first, to our knowledge, reliable archaeological window into the Southeast Asian colonization of Madagascar. They additionally suggest that initial Southeast Asian settlement in Africa was not limited to Madagascar, but also extended to the Comoros. Archaeobotanical data may support a model of indirect Austronesian colonization of Madagascar from the Comoros and/or elsewhere in eastern Africa.
Alison Crowther, et al., "Ancient crops provide first archaeological signature of the westward Austronesian expansion" 113(24) PNAS 6635-6640 (2017) doi: 10.1073/pnas.1522714113
From the introductory body text:
From the introductory body text:
One line of evidence that has been largely overlooked in archaeological investigations of Madagascar and, indeed, eastern Africa more broadly is ancient plants. However, it is estimated that some 10% of Madagascar’s flora was introduced from elsewhere, and plant introductions include a significant number of staple crops, spices, and arable weeds of Asian origin. Historically or currently important crops on Madagascar, like banana (Musa spp.), yam (Dioscorea alata), taro (Colocasia esculenta), and coconut (Cocos nucifera), are Southeast Asian cultivars. Asian rice (Oryza sativa), which was domesticated separately in East and South Asia but is the basis of traditional agriculture across much of Madagascar today, was also widely grown in Southeast Asia by the first millennium CE. Other Asian crops, like mung bean (Vigna radiata) and Asian cotton (Gossypium arboreum), are also cultivated on Madagascar. The fact that early crop introductions to Madagascar may have arrived with Austronesian settlers seems particularly feasible given that Austronesian expansion into the Pacific was linked to the spread of a similar suite of cultivars.
To directly explore early cultivated plants on Madagascar and their potential to inform on its colonization history, we collected new archaeobotanical data from the island as well as contemporaneous sites on the African mainland coast (Kenya and Tanzania) and nearshore islands (Pemba, Zanzibar, and Mafia) and the Comoros. These data were collected from 18 sites in total, dating between approximately 650 and 1200 calibrated years (cal) CE. The archaeobotanical datasets derive primarily from recent excavations at 16 sites, during which systematic sampling for charred macrobotanical remains at high stratigraphic resolution was conducted. They are supplemented by existing records from one of the sites (Sima) as well as data from previous excavations at two other sites in the Comoros. The combined dataset includes 2,443 identified crop remains recovered from >7,430 L sediment across the sites and is supported by 48 accelerator MS (AMS) radiocarbon dates, 43 of which were obtained directly on crop seeds.From the conclusion:
Although the presence of Asian crops that likely originate from Southeast Asia on early sites in Madagascar corresponds well with linguistic, genetic, and ethnographic evidence for a prehistoric migration of people from this region, the finding that these crops also dominate early assemblages on the Comoros is rather unexpected. In particular, the presence of Asian crops at sites in the Comoros earlier than at sites on Madagascar is of significant interest, and although sampling and preservation biases cannot be discounted, may reflect Austronesian colonization of the Comoros before Madagascar. As noted, however, Comorians today speak Bantu languages, and in addition, preliminary molecular genetic studies suggest that they possess only a small proportion of Southeast Asian ancestry. Nonetheless, the population of the Comoros is small and has been historically subject to significant population bottlenecks and Bantu input as a result of slave raiding and trading over many centuries. Thus, it is possible that the Comoros were settled at an early date by a Southeast Asian population that was later genetically and linguistically swamped.
Direct colonization from Southeast Asia is common to many models of Madagascar’s Austronesian settlement, particularly those put forward by archaeologists and geneticists. However, linguistics have offered another perspective, with some linguists taking the view that the remarkable unity of Bantu loanwords and grammatical features throughout Malagasy dialects can only be explained through initial Austronesian settlement on the African mainland and/or the Comoros. Early Southeast Asian presence or influence on the Comoros has also been suggested on the basis of the apparent presence of several 10th or 11th century “Austronesian-type” furnaces on Mayotte as well as findings of shell-impressed pottery at early sites on the islands. These suggested Austronesian linkages, however, have been both limited and contentious. This study suggests that they deserve reinvestigation together with the argument that the Comoros may have served as a key base for Southeast Asian commercial activity in the western Indian Ocean, including an alternative slave-trading corridor. Independent linguistic, genetic, and archaeological studies are required to examine the role of the Comoros in early Indian Ocean population movements and commercial trade.
